|
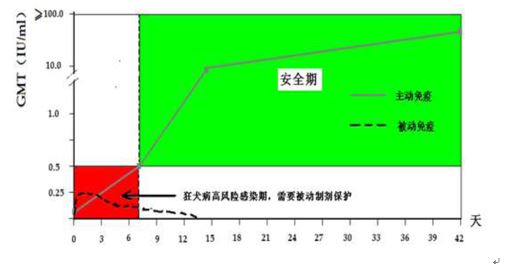
发布时间:2018-12-25 一、概述 狂犬病是由狂犬病病毒感染所致的一种动物源性传染病,病死率几乎100%,据原卫计委统计数据,近15年我国狂犬病年发病死亡人数在600~3300例。2004-2014年,狂犬病死亡人数一直高居我国传染病死亡数的前3位[1]。全国年暴露人口数逾4000万,接种疫苗人数为1500万左右[2],是我国重要的公共卫生威胁。 根据我国《狂犬病预防控制技术指南(2016年版)》[3],目前对已经出现疾病症状的狂犬病没有公认有效的治疗方法,只能预防。在我国,暴露后免疫比暴露前预防的成本效益更高,成为主要的预防方式。暴露后立刻给予有效的狂犬病疫苗和抗体,能够使人体产生对狂犬病的免疫力。世界卫生组织(WHO)狂犬病专家咨询委员会建议[4, 5],对于狂犬病病毒III级暴露者,应在接种疫苗的同时对伤口进行彻底清洗并在周围浸润注射被动免疫制剂,即人狂犬病免疫球蛋白(human rabies immune globulin,HRIG)或马源抗狂犬病血清(Equine rabies antiserum,ERA),以阻止病毒进入神经组织从而获得快速保护作用。对于免疫功能严重低下的暴露者,即使II 级暴露,也应联用被动免疫制剂。被动免疫制剂的机制为在第一针疫苗注射后至机体产生足量抗体之前的窗口期提供即时的免疫保护。但上述被动免疫制剂供应量有限,价格偏高,有血清病类过敏反应和血源传播疾病的潜在风险。 狂犬病单克隆抗体因批间效价差异小、安全性提高、可大量制备等优势,有替代HRIG、ERA的潜力,成为该领域的研发热点[6]。目前国际上已有数家公司投入到基因工程重组抗狂犬病毒单抗的研发领域:印度血清研究所开发的SII RMab于2016年在印度获批上市;荷兰Crucell公司、印度Zydus研究中心以及中国兴盟生物科技、中国华北制药的相关产品均已进入II、III期临床研究。 西欧、加拿大、美国、日本等发达国家因在动物免疫、管理方面的重视,已基本消除了犬狂犬病,在这些国家狂犬病通常被列为罕见病。而99%的人间狂犬病发生在发展中国家,中国狂犬病发病仅次于疫情最严重的印度,狂犬病治疗用药在我国有广大的目标人群,因此新药临床试验的实施及评价不应与欧美日等发达国家标准完全一致。由于狂犬病一旦发病,临床结局为死亡,作为被动免疫预防性用药临床试验有诸多安全风险和伦理问题,在新药临床试验设计时应对研究人群、用法用量、研究主要终点、联合用药、研究周期等慎重考虑。本文对狂犬病病毒单克隆抗体新药临床试验设计和评价重点关注问题进行了讨论分析。 二、临床试验的重点考虑 (一)受试者 1.早期和探索性试验受试者选择 由于狂犬病临床结局的特殊性,其预防用药的有效性也是安全性评价的重要组成部分。出于伦理和临床研发考虑,早期和探索性试验建议选择无狂犬病暴露、无接种史的健康受试者。同时,由于早期试验以抗体滴度为疗效指标,健康志愿者也能满足疗效观察的需求。 2.确证性试验受试者选择 在确证性试验阶段,应考虑受试者与临床实际用药人群的一致性。狂犬病单克隆抗体出于暴露后免疫(PEP)的研发目的,应选择狂犬病毒暴露且需要应用被动免疫制剂者,其暴露定义应符合当前学术界共识。参考当前狂犬病防控技术指南设定和临床研发实际,建议确证性试验受试者选择免疫功能正常的III级暴露者。 具体制定受试者选择标准时,建议对暴露动物和暴露身体部位进行规定,并在试验中记录具体情况。犬是我国狂犬病的主要传染源,其次是猫,占95%以上,狐、狼、豺、鼬獾、貉、臭鼬、浣熊、猫鼬和蝙蝠等是狂犬病的自然储存宿主,可感染狂犬病病毒成为传染源。暴露于蝙蝠均属于III 级暴露。禽类、鱼类、昆虫类、爬行类(如蜥蜴、蛇、 龟鳖)等不感染和传播狂犬病,啮齿类和兔形目极少感染狂犬病,也未发现此类可分为导致人间狂犬病的证据。 狂犬病潜伏期长短与病毒的毒力、侵入部位的神经分布等因素相关,病毒数量越多、毒力越强、侵入部位神经越丰富、越靠近中枢神经系统,潜伏期越短[4, 5]。头面部、手指、手臂、会阴部等神经终板丰富的部位暴露比其他部位暴露,多处暴露比单处暴露更高危。在募集受试者时,建议尽可能控制创面的大小、深度等,因外伤的复杂程度不同,暴露者还可能患严重并发症以及继发的细菌感染,应保证组间研究基线一致。 对全部咬伤暴露人群的动物进行病毒学检测确定高危人群的难度较大,可在临床研究中对一定比例的感染源进行病毒学检测。狂犬病潜伏期无任何诊断方法,因此无需对受试者进行病毒学检测。 被动免疫制剂应尽早使用,最好在伤口清洗完成后立刻开始。疫苗应用7 天后其主动免疫应答反应已经出现,此时再使用被动免疫制剂意义不大(见图)。临床试验中,为获得理想状态和结果,应对暴露后被动免疫时机进行规定,建议一般为24小时之内。
图. 狂犬病被动免疫作用机制简图[3, 7] 根据WHO的统计数据,40%被狂犬咬伤者为15岁以下的儿童。因此鼓励在新药研究中纳入儿童受试者为临床用于儿童提供依据,建议在确证成人应用的安全有效性之后,再考虑儿童的试验研究。 完成免疫功能正常暴露人群研究后,在有证据可以预测新药可能在免疫功能抑制者、合并HIV感染者、肝肾功能失代偿者等人群中的安全有效性的情况下,才可进行相应人群的临床试验。 (二)终点指标 因研究药物为狂犬病预防性用药,一旦发病临床结局为死亡,因此以疾病的转归和结局作为唯一主要研究终点存在困难和伦理问题,可以选择与理想主要终点最相关的疗效指标作为临床试验的主要评价指标。 1.血清学指标 一般选择狂犬病毒中和抗体(Rabies Virus Neutralizing Abs,RVNA)监测免疫效果。WHO推荐抗狂犬病病毒中和抗体标准检测方法包括快速荧光灶抑制试验(Rapid Fluorescent Focus Inhibition Test,RFFIT)和小鼠脑内中和试验(Mouse Neutralization Test,MNT),RFFIT 方法也是我国现行药典规定的检测狂犬病病毒中和抗体的标准方法之一。目前,被动免疫尚无RVNA有效阈值水平的推荐。对于狂犬病疫苗,WHO认为狂犬病中和抗体水平等于或高于0.5IU/ml时,接种者才具备有效的保护能力。美国免疫接种咨询委员会(ACIP)推荐抗体有效水平为RFFIT法血浆稀释5倍,即0.1IU/mL。考虑到新药研发为预防性目的,一种剂量需覆盖所有病毒株、各病毒暴露水平、各类人群,结合推荐RVNA水平推算过程,可以考虑以WHO推荐的RVNA≥0.5IU/ml作为临床试验中被动免疫有效水平。但应注意到狂犬病单克隆抗体为伤口局部和全身中和作用,血清抗体水平是替代药效学指标,且与疫苗联用时测得RVNA是单克隆抗体与疫苗主动免疫的叠加效果。建议进一步进行研究并采集数据,以更进一步支持血清学指标的科学性与合理性。 新药最终是与狂犬病疫苗联用,而疫苗一般在接种后7~14天才能使机体出现足量抗体。因此,建议在第0天用药前进行第一次RVNA检测,观察期间还需多次检测RVNA,注射新药后7天内至少进行1次RVNA检测,窗口期内检测时机应恰当,太早可能不能达到抗体有效水平,太晚不能合理说明新药在窗口期的被动免疫作用。 2.临床结局 狂犬病潜伏期一般为1-3个月,极少数短至两周以内或长至一年以上,此期间无任何其他确诊方法。一旦发病,通常在临床症状出现后7-8天即到发病晚期,死亡率近100%。建议临床结局观察时间至少1年。 3.其他 目前的被动免疫制剂在伤口周围浸润注射,以在高风险感染期使伤口局部获得高浓度的中和抗体,阻断病毒在伤口中扩散,但尚无公认有效方法检测伤口处局部中和效果,血浆的中和抗体水平不能说明伤口处的抗体浓度,新的检测方法及效果仍有待探索,鼓励在符合伦理的前提下在临床研究中展开探索。 4.主要终点及次要终点的选择 临床试验主要终点指标一般可以考虑选择用药后第7天或第14天的RVNA≥0.5IU/ml的相对抗体阳性率或RVNA几何平均滴度,具体选择时应考虑到终点指标对被动免疫效果的说明性,以及试验设计、阳性对照情况。 对于次要终点,早期临床研究至少需包括:用药后第7天、第14天和其他多个不同时点的RVNA几何平均滴度及相对抗体阳性率。疗效确证性研究的次要终点还需包括受试人群的狂犬病发病率和生存率(临床结局),上市前至少应提供3个月的观察数据,如临床试验显示新药的有效性和安全性良好,经监管机构认可后,临床结局的长期观察(至少1年)可在有条件上市后提供。 (三)用法用量 被动免疫为在疫苗诱导产生有效抗体前,迅速短暂的提供病毒中和抗体,而疫苗诱导产生的抗体可持续存在数年,因此其尚不能取代疫苗,应与疫苗联合应用。局部伤口的浸润注射对于使用疫苗后机体主动免疫产生的影响较小,狂犬病单克隆抗体与HRIG作机制相同,其用法也应为伤口浸润注射,参考相关指南HRIG推荐用法设置。 新药的用量需经过探索,一方面剂量不能过低,以保证免疫的有效性和对一般人群的安全性;另一方面考虑到,使用剂量过高会过度干扰疫苗的主动免疫保护。 (四)安全性评价 在实际情况下,临床应用存在部分非狂犬病毒暴露人群也可能使用被动免疫制剂,故该类产品的安全性尤其需要关注。因狂犬病适应症临床结局的特殊性,新药的有效性(是否干扰疫苗、是否广泛覆盖狂犬病毒株)也是安全性评价的重要内容。 需要观察的内容主要包括生命体征、体格检查、常规的实验室检查、心电图、胸片、腹部B超、全身反应、局部反应等。因品种特点有特殊考虑的,也需要考虑到相应的安全性风险,并在临床试验中观察。单克隆抗体应关注过敏反应、流感样症状、胃肠道症状、低血压等,结合品种临床前研究和同类品种的研发经验,对潜在风险设计风险控制措施。同时,大分子药物需要在临床试验中设计观察免疫原性,是否有抗抗体产生。新药的早期安全性观察至少42天,确证性临床研究安全性观察至少1年。 三、不同开发阶段的试验设计要点 (一)新药进入临床试验的基本条件 新药进入临床试验前,应进行充分的药效、药代动力学和毒理学等非临床研究。为人体临床试验方案设计提供参考和支持,如起始剂量、给药间隔、停药标准、主要安全性风险及风险控制措施等。 狂犬病病毒单克隆抗体主要在暴露局部及全身起中和作用,而临床试验中检测伤口处局部中和效果困难,非临床药效学研究中建议包括动物局部组织分布试验。WHO建议[8],抗狂犬病单克隆抗体制剂应将针对病毒不同抗原位点的多株单抗组合成“鸡尾酒式”组合制剂,以保证单抗制剂对不同病毒株或病毒的不同基因型的有效性。因此,临床前需明确新药中和毒株能够覆盖我国流行的毒株,且安全有效,方可进入临床试验。 (二)早期临床试验 早期研究应该包括人体耐受性研究、人体药代动力学(PK)、药动学/药效学(PK/PD)研究和药物相互作用研究,研究目的是评价药物的人体耐受剂量范围、PK特征和暴露/效应关系,为后期研究给药方案提供支持。 1.人体耐受性研究 狂犬病被动免疫为短期用药,药效保持时间应至少为7天,一般不超过2周,因此通常狂犬病单克隆抗体应为只注射一次的长效制剂,如新药临床前研究数据显示无需多次给药,可只进行单次给药耐受性试验。试验建议在无狂犬病暴露、无接种史的健康受试者中进行,可以是开放、基线对照的,也可以采用随机、盲法和安慰剂对照方法以提高观察结果的有效性。 2.药代动力学和药动学/药效学(PK/PD)研究 应充分说明在预期剂量范围内的药代动力学特性,如是否呈现线性药代动力学特征、药代动力学特征个体间变异,及造成变异的影响因素等。单抗类药物体内清除途径通常与小分子的代谢途径不同,蛋白质水解作用和靶点介导的清除机制在该类药物消除过程中通常发挥重要作用。 健康受试者和暴露者体内药代动力学特征可能存在差异,故应在狂犬病暴露者开展必要的人体药代动力学研究。在早期临床开发阶段,选择合适的药代动力学模型,以RVNA作为药效动力学指标,进行药动学/药效学评估,可更全面了解药物的暴露/效应作用特点。药效动力学效应指标应满足多点采样要求。结合后期临床研究,开展必要的群体药代动力学研究,以寻找药代动力学的影响因素,如性别,体重,暴露的伤口大小、深度、位置等。 3.药物相互作用研究 狂犬病单克隆抗体通常与狂犬病疫苗联合应用,应考察联用的耐受性、药代动力学和药效学。选择无狂犬病暴露、无接种史的健康受试者,设置对照研究,分组应至少包括新药组、疫苗组、新药+疫苗组,可选择盲法。 另外,建议根据前期非临床研究等考虑新药是否可能与其他药物存在相互作用,并进行相应研究。 (三)探索性试验 探索性临床试验阶段的主要目的是初步收集新药有效性和安全性数据,为疗效确证临床试验中给药剂量和方案提供依据。该阶段多采用随机、对照试验设计。受试者可选择无狂犬病暴露、无接种史的健康受试者,主要终点选择用药后第7天或第14天的RVNA≥0.5IU/ml的相对抗体阳性率或RVNA几何平均滴度,并同时观察药物安全性。试验药物设置多个剂量组,对照药应选择目前现有的标准狂犬病被动免疫药物,基于综合因素考虑,优先推荐HRIG作为阳性对照,通过与HRIG比较确定新药的剂量。探索性试验应合并使用疫苗探索,即不同剂量新药+疫苗与对照药+疫苗的对照研究,在开始用药后建议至少观察42天。 早期和探索性研究可灵活设计。如设置的受试者、研究主要终点等一致,在保证研究质量和样本量的前提下,可以选择性与部分I期临床试验合并研究,合理利用临床试验资源,提高研发效率。 (四)疗效确证性试验 疗效确证性试验目的在于在探索性试验基础上、在目标人群进一步确证临床获益情况,为新药上市提供充分数据。受试者一般选择免疫功能正常的WHO规定的III级暴露者。由于暴露后预防失败的后果是死亡,因此前期需有足够有效性数据支持,提交前期临床研究数据及可行的疗效确证性试验方案,经监管机构审核认可后,方可进入疗效确证性试验。通常采用随机、对照、双盲的试验设计,优先推荐HRIG作为阳性对照,进行新药+疫苗与HRIG+疫苗的对比研究。主要终点一般选择用药后第7天或第14天的RVNA≥0.5IU/ml的相对抗体阳性率或RVNA几何平均滴度,临床结局(受试者发病和生存情况)作为次要终点,建议上市前至少观察3个月,经监管机构认可后,长期临床结局观察(至少1年)可在有条件上市后进行。 试验一般推荐非劣效设计。优效设计试验获得阳性结果难度大,需要大样本量。选择非劣效设计,其非劣效界值(margin)的确定至关重要,若界值太大,会把疗效远不如对照药的药物判断为有效或等效,若界值太小,则可能误判为无效而不能及时上市。建议基于前期研究数据和历史文献,合理确定非劣效界值。应根据试验设计的选择和统计学方法,进行样本量的计算。 (五)试验的质量控制 在狂犬病临床试验中需注意临床试验的质量控制,包括但不限于: 1.机构选择 我国狂犬病多发于农村,暴露者被动物咬伤后,一般会就近处理而选择医院急诊或区域卫生防疫站,因此临床试验机构选择需注意。 2.治疗标准化及培训 暴露后预防处置首先要尽早进行伤口局部处理,包括对每处伤口进行彻底的冲洗、消毒以及后续的外科处置,这对于预防狂犬病发生、避免继发细菌感染具有重要意义,临床试验方案中应规定一套标准作业程序(SOP)。狂犬病单克隆抗体的用法为伤口浸润注射,每个受试者情况一般会有区别,每个研究人员操作主观差异性大,因此也需要详细的治疗SOP,并对研究者进行培训。
参考文献
[1]Yao HW, Yang Y, Liu K, et al. The spatiotemporal expansion of human rabies and its probable explanation in mainland China, 2004-2013[J]. PLoS Negl Trop Dis, 2015, 9(2): e0003502. [2]Yin CP, Zhou H, Wu H, et al. Analysis on factors related to rabies epidemic in China from 2007-2011[J]. Virol Sin, 2012, 27(2): 132-43. [3]周航. 狂犬病预防控制技术指南(2016版)[J]. 中华流行病学杂志, 2016, 2(37). [4]WHO Expert Consultation on Rabies. Second report[J]. World Health Organization technical report series, 2013, 982. [5]Organization WH. Rabies vaccines. WHO position paper[J]. Wkly Epidemiol Rec, 2018, 16(93): 201-220. [6]赖永洁. 人用狂犬病疫苗和被动免疫制剂研发进展[J]. 中国人兽共患病学报, 2018, 6(34). [7]俞永新主编. 狂犬病和狂犬病疫苗[M]. 北京:中国医药科技出版社, 2009: [8]WHO Consultation on a Rabies Monoclonal Antibody Cocktail for Rabies Post Exposure Treatment. WHO. . |
友情链接
联系方式
 010-64929757
010-64929757  service@gcpunion.org
service@gcpunion.org  北京市北三环中路朝阳区安外小关北里43号渔阳置业大厦A座706
北京市北三环中路朝阳区安外小关北里43号渔阳置业大厦A座706 
微信公众号
微博公众号
